李劲松、胡苹合作利用“人造精子细胞”介导半克隆技术构建出新型Ⅰ型强直性肌营养不良症小鼠模型
来源:
时间:2020-02-11
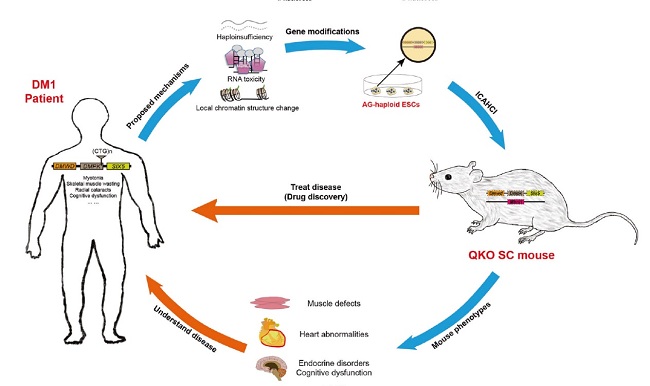
2月5日,中国科学院分子细胞科学卓越创新中心(生物化学与细胞生物学研究所)李劲松研究组、胡苹研究组合作的文章“Dosage effect of multiple genes accounts for multisystem disorder of myotonic dystrophy type 1”在国际学术期刊《细胞研究》上作为封面文章发表。该研究利用“人造精子细胞”介导的半克隆技术,快速构建多基因杂合突变小鼠模拟人Ⅰ型强直性肌营养不良症(DM1)的多基因剂量不足效应,成功地在小鼠中再现了DM1的大部分病症,为开展DM1致病机制和药物筛选研究提供了新模型。
人I型强直性肌营养不良症(DM1)是一种遗传性疾病,是由肌营养不良症肌强直蛋白激酶(DMPK)基因3’UTR区CTG重复序列的异常扩增引起的复杂疾病。正常人的CTG短串联重复序列平均为37个,DM1患者的CTG超过50个。DM1严重程度和发病年龄与CTG的数量有关,主要症状包括肌强直、肌萎缩、肌无力、心脏缺陷、白内障等。随着重复序列的异常增加(~4000),可产生新生儿呼吸衰竭,心脏发育不全等,甚至造成新生儿致死(先天性DM1, CDM)。重复序列的异常增加不仅影响DMPK的mRNA和蛋白水平(基因单倍剂量不足效应),同时也改变了邻近染色质的结构并降低SIX5和DMWD的表达(局部染色质结构改变效应)。此外,许多研究还表明异常延迟的DMPK mRNA会滞留在核内,并募集了剪接调控因子,如MBNL1,在核内形成核糖核聚集体(病灶),引起RNA毒性(RNA毒性效应)。然而,单基因纯合突变小鼠模型(包括Dmpk、Six5和Mbnl1)均不能模拟DM1患者的大部分症状,更不能模拟出CDM1的症状,提示重复序列的异常扩增导致多基因的表达的同时下降可能是DM1复杂病症产生的原因,但是到目前为止仍然缺乏直接的实验证据。
为了验证这一假说,研究团队利用李劲松课题组建立的孤雄单倍体胚胎干细胞(AG-haESCs)能够替代精子产生小鼠的半克隆技术(又称为“人造精子细胞”介导半克隆技术),结合CRISPR/Cas9基因编辑技术,成功构建了三基因(Dmpk, Six5, Mbnl1)敲除的“人造精子细胞”系,并通过卵子注射一步获得携带三基因杂合敲除的半克隆小鼠(Dmpk+/-; Six5+/-; Mbnl1+/-)。表型分析发现该小鼠产生了典型的DM1表型,包括四月龄时出现的肌肉无力、肌肉萎缩、严重的运动障碍;小肠和膈肌结构异常(消化和呼吸功能障碍); 12个月龄小鼠出现心肌纤维结构异常(心脏功能障碍)等。
三基因杂合敲除的小鼠可以作为一种新的DM1模型,模拟DM1成年患者中观察到的大多数病理表型,说明DM1中出现的病理表型的确是由多个基因剂量下降协同产生的。然而,该小鼠没有出现CDM的表型,提示其它基因的剂量变化可能与此有关。为此,研究人员进一步构建了Dmwd+/-半克隆小鼠(Dmpk的上游基因,有报道显示患者中DMWD的表达水平下降),表型分析发现该小鼠出现肌肉萎缩的表型,说明Dmwd的确与DM1的表型有关。
团队进一步将带有四基因(Dmpk, Six5, Mbnl1, Dmwd)敲除的“人造精子细胞”注射到卵子中产生了四基因杂合敲除的小鼠(Dmpk+/-; Six5+/-; Mbnl1+/-; Dmwd+/-)。与三基因杂合敲除的小鼠相比,四基因杂合敲除的半克隆小鼠呈现出先天性DM1表型:出生后几个小时内就出现大约22%的死亡,肺扩张失败是导致四基因杂合敲除半克隆幼仔出生后死亡的主要原因,而导致肺扩张失败的原因可能是膈肌发育异常;存活下来的小鼠出现严重的肌强直、肌无力、肌肉萎缩和运动缺陷;部分小鼠出现严重的肠道异常和心脏发育异常,约46%小鼠在断奶前死亡;剩下的小鼠可以发育至成年,呈现出典型的DM1表型,包括白内障等。
进一步研究发现三基因和四基因杂合敲除小鼠的肌肉干细胞数量以及增殖能力与对照无差异,然而其干性降低,从而导致了肌肉分化的缺陷。利用这一特点,研究人员建立了促进肌肉干细胞分化的小分子筛选系统,并发现了一种在细胞水平上能缓解肌肉分化异常表型的小分子化合物,为进一步开展大规模DM1药物筛选研究奠定了基础。
该研究通过利用“人造精子细胞”介导半克隆技术快速构建多基因剂量下调小鼠模型模拟出DM1的多数病症,提供了直接证据支持CTG重复序列的异常扩增导致多个基因表达水平变化(下调)从而诱发DM1的复杂病理表型的假说;同时,这些模型的建立为深入研究DM1的发病机理和进行药物筛选奠定了基础。
美国贝勒医学院人类遗传学家Richard H. Finnell教授在同期刊发了评论文章,认为该研究为人强直性肌营养不良症研究和治疗提供了可靠的小鼠模型,解决了该领域长期缺乏有效小鼠模型的困境;肯定了半克隆技术结合CRISPR/Cas9基因编辑技术产生多基因杂合突变小鼠模型的高效性及有效性;并认为此技术有望成为包括出生缺陷、衰老、心血管、癌症、神经发育相关疾病在内复杂型疾病的一个强有力研究工具。
尹奇、王红叶、李钠、丁一夫、谢振飞和金立方为本文的共同第一作者,李劲松研究员和胡苹研究员为共同通讯作者。本工作得到了中国科学院分子细胞科学卓越创新中心(生物化学与细胞生物学研究所)鲍岚研究员、周斌研究员、福建医科大学陈万金教授的大力支持,该研究受到了分子细胞中心化学生物学技术平台、细胞分析技术平台、实验动物技术平台和基因组标签计划研发中心的大力支持,同时还得到国家自然科学基金委、国家科技部、上海市科委和中国科学院的经费资助。
DM1小鼠模型的构建及其潜在应用
