2021年11月2日,国际学术期刊Cell Research在线发表了中国科学院分子细胞科学卓越创新中心(生物化学与细胞生物学研究所)孙丽明研究组与上海科技大学生命科学与技术学院王华翌课题组合作的研究成果“Spontaneous necroptosis and autoinflammation are blocked by an inhibitory phosphorylation on MLKL during neonatal development”。该研究首次报道了细胞坏死执行蛋白MLKL存在一个物种间保守的抑制性磷酸化修饰,通过阻断MLKL的膜转运抑制细胞坏死的发生,并在小鼠早期发育过程中扮演着重要的炎症损伤保护作用。
程序性细胞坏死 (Necroptosis) 是一种“膜裂解性”程序性细胞死亡方式。其典型的特点是细胞膜破裂、细胞内容物释放。过度的细胞坏死引起周围组织的炎症反应,其参与癌症、炎症性疾病、感染性疾病等众多病理过程。因此细胞坏死在正常的机体组织中是被严格调控的。但目前科学家对于其负调控机制还知之甚少。
细胞坏死可以被炎症因子、病原相关分子模式(PAMP)等多种因素触发。其关键激活节点是激酶RIP3的活化和其对坏死执行蛋白MLKL的磷酸化激活。被RIP3磷酸化的MLKL继而发生寡聚化并转位到细胞膜,直接造成细胞膜损坏,最终导致细胞的坏死。
通过细胞内稳定同位素标记(SILAC)的质谱分析,研究人员收集诱导程序性细胞坏死发生后的幸存细胞,发现人源MLKL的83位点丝氨酸(S83)的磷酸化被富集。细胞存活性实验验证发现S83的磷酸化抑制MLKL介导的细胞坏死。进一步的细胞生物学及生物化学实验证明,S83的磷酸化不依赖于RIP3且不影响上游RIP3对MLKL的C端造成的磷酸化激活,而是通过直接抑制MLKL的N端寡聚化,进而阻止MLKL转位到细胞膜造成细胞坏死。
通过序列比对,研究人员发现对应人源MLKL的83丝氨酸的氨基酸位点在哺乳动物中高度保守。在同源位点磷酸化模拟突变(MlklS82E/S82E)的小鼠中,其真皮成纤维细胞MDF和骨髓来源的巨噬细胞BMDM的程序性坏死被阻断,证明小鼠中该位点(S82)的磷酸化修饰同样具有抑制细胞坏死的功能。此外,蛙皮素诱导的细胞程序性坏死通路参与的胰腺炎被抑制。有趣的是,在S82位点磷酸化功能缺失性突变的小鼠中(MlklS82G/S82G),其纯合子后代有大约15%的幼仔在哺乳期陆续死亡,并伴有肺、肝脏等多器官的严重炎症损伤,以及程序性细胞坏死标记物p-MLKL (S345) 上调。这说明鼠源MLKL S82位点的磷酸化,在小鼠出生后发育过程中,阻止了自发性程序性细胞坏死造成的炎性损伤。此外,存活下来的小鼠的多个器官中也检测到轻微的炎症和细胞坏死,随着发育到成年,这些小鼠MLKL的mRNA水平和蛋白水平显著下降且多器官炎症消失,提示细胞坏死过度激活的小鼠中可能存在转录和翻译水平上的调控机制来减少细胞坏死的发生。
该研究报道了MLKL (S83/82位点)的抑制性磷酸化修饰,证明其作为程序性细胞坏死在执行膜裂解之前的重要检查点(checkpoint),保护机体出生后由于过度的细胞坏死造成的炎性损伤而发挥重要的作用。
分子细胞卓越中心孙丽明研究员和上海科技大学王华翌研究员为该论文的共同通讯作者,该工作得到国家自然基金委和中科院先导科技专项(A类)的经费资助。
文章链接:https://www.nature.com/articles/s41422-021-00583-w
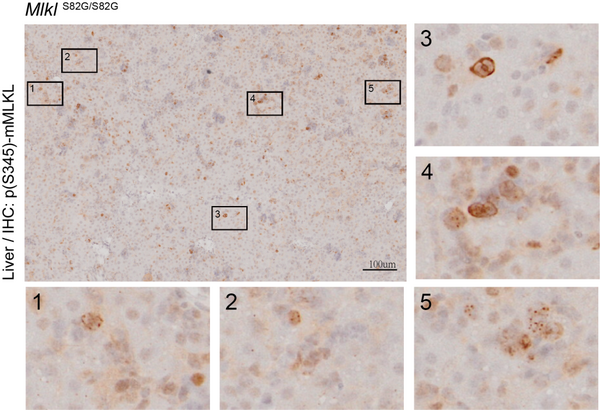
MLKL抑制性磷酸化功能缺失突变(MlklS82G/S82G)小鼠肝脏发生程序性细胞坏死。免疫组化显示肝细胞膜上出现被RIP3激活的MLKL(pS345)信号。